The io module
The io module enables loading of different input formats and conversion to different output formats.
Currently the following input/output formats are supported:
Input |
Output |
|---|---|
mol2 (file, text) |
|
pdb (file, text) |
|
mol2 (file, text) |
|
pdb (file) |
|
[1]:
%load_ext autoreload
%autoreload 2
[2]:
from opencadd.io.dataframe import DataFrame
from opencadd.io.rdkit import Rdkit
from opencadd.io.biopython import Biopython
[3]:
from pathlib import Path
DATA_PATH = Path("../../opencadd/tests/data/io")
[4]:
from rdkit.Chem import Draw
from rdkit.Chem.Draw import IPythonConsole
Load structural data as DataFrame
Return data from different input formats as DataFrame with the following default columns:
atom.id: Atom serial number.atom.name: Atom name.atom.x,atom.y,atom.z: Orthogonal coordinates for X, Y, Z in Angstroms.atom.charge: Atom charge.residue.pdb_id: Residue PDB ID.residue.name: Residue name.
It is also possible to return a DataFrame in the verbose version, that contains additional columns available in the input format (but not in other input formats).
Columns available from the mol2 format only are:
atom.type: SYBYL atom type set in the mol2 format.residue.subst_id: ID number of the substructure containing the atom.residue.subst_name: Name of the substructure containing the atom: Residue name + residue PDB ID.
Columns available in the pdb format only are:
record.name: Record name set in the pdb format, i.e.ATOMorHETATM.atom.symbol: Atom symbol.atom.occupancy: Atom occupancy.atom.bfactor: Atom B factor.atom.alternative_model: Alternative atom positions.structure.chain: Structure chain.
From the mol2 format
Check out the mol2 format here.
From mol2 files: Return a mol2 file’s content as DataFrame (mol2 files can have 9 (default) or 10 columns).
[5]:
# Mol2 files with 10 columns
DataFrame.from_file(DATA_PATH / "2itz_chainA_protein.mol2")
[5]:
| atom.id | atom.name | atom.x | atom.y | atom.z | residue.id | residue.name | |
|---|---|---|---|---|---|---|---|
| 0 | 1 | N | 3.5231 | 35.933102 | 15.596400 | 697 | GLU |
| 1 | 2 | H1 | 3.0492 | 36.782101 | 15.323000 | 697 | GLU |
| 2 | 3 | H2 | 3.1546 | 35.156300 | 15.066500 | 697 | GLU |
| 3 | 4 | H3 | 4.5121 | 36.026001 | 15.414000 | 697 | GLU |
| 4 | 5 | CA | 3.3077 | 35.690701 | 17.044600 | 697 | GLU |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 4876 | 4877 | HD2 | 3.5410 | 42.831100 | 42.216801 | 1019 | PRO |
| 4877 | 4878 | HD3 | 1.9924 | 43.615398 | 42.610600 | 1019 | PRO |
| 4878 | 4879 | N | 3.6128 | 43.025902 | 38.110600 | 1020 | GLN |
| 4879 | 4880 | H1 | 4.0174 | 42.840500 | 37.203999 | 1020 | GLN |
| 4880 | 4881 | H2 | 4.1575 | 42.873901 | 38.947399 | 1020 | GLN |
4881 rows × 7 columns
[6]:
# Mol2 files with 9 columns
DataFrame.from_file(DATA_PATH / "2itz_protein.mol2")
[6]:
| atom.id | atom.name | atom.x | atom.y | atom.z | residue.id | residue.name | |
|---|---|---|---|---|---|---|---|
| 0 | 1 | C | -68.010002 | -5.498000 | -49.028000 | 697 | GLU |
| 1 | 2 | O | -67.916000 | -4.916000 | -50.118999 | 697 | GLU |
| 2 | 3 | CA | -67.651001 | -6.991000 | -48.900002 | 697 | GLU |
| 3 | 4 | N | -67.939003 | -7.787000 | -50.118999 | 697 | GLU |
| 4 | 5 | CB | -66.192001 | -7.196000 | -48.494999 | 697 | GLU |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 4878 | 4879 | H1 | -55.506401 | -10.457300 | -32.785599 | 3035 | HOH |
| 4879 | 4880 | H2 | -55.441399 | -9.182800 | -31.851601 | 3035 | HOH |
| 4880 | 4881 | O | -58.311001 | -15.659000 | -30.250999 | 3045 | HOH |
| 4881 | 4882 | H1 | -58.885601 | -15.679400 | -31.069201 | 3045 | HOH |
| 4882 | 4883 | H2 | -57.761101 | -16.492201 | -30.310301 | 3045 | HOH |
4883 rows × 7 columns
In order to get all columns from the input, set verbose=True.
[7]:
DataFrame.from_file(DATA_PATH / "2itz_chainA_protein.mol2", verbose=True)
[7]:
| atom.id | atom.name | atom.x | atom.y | atom.z | residue.id | residue.name | atom.type | residue.subst_id | residue.subst_name | atom.charge | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | N | 3.5231 | 35.933102 | 15.596400 | 697 | GLU | N.4 | 1 | GLU697 | 1.0 |
| 1 | 2 | H1 | 3.0492 | 36.782101 | 15.323000 | 697 | GLU | H | 1 | GLU697 | 0.0 |
| 2 | 3 | H2 | 3.1546 | 35.156300 | 15.066500 | 697 | GLU | H | 1 | GLU697 | 0.0 |
| 3 | 4 | H3 | 4.5121 | 36.026001 | 15.414000 | 697 | GLU | H | 1 | GLU697 | 0.0 |
| 4 | 5 | CA | 3.3077 | 35.690701 | 17.044600 | 697 | GLU | C.3 | 1 | GLU697 | 0.0 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 4876 | 4877 | HD2 | 3.5410 | 42.831100 | 42.216801 | 1019 | PRO | H | 302 | PRO1019 | 0.0 |
| 4877 | 4878 | HD3 | 1.9924 | 43.615398 | 42.610600 | 1019 | PRO | H | 302 | PRO1019 | 0.0 |
| 4878 | 4879 | N | 3.6128 | 43.025902 | 38.110600 | 1020 | GLN | N.am | 303 | GLN1020 | 0.0 |
| 4879 | 4880 | H1 | 4.0174 | 42.840500 | 37.203999 | 1020 | GLN | H | 303 | GLN1020 | 0.0 |
| 4880 | 4881 | H2 | 4.1575 | 42.873901 | 38.947399 | 1020 | GLN | H | 303 | GLN1020 | 0.0 |
4881 rows × 11 columns
From mol2 text: Return a mol2 string (text) as DataFrame. This functionality is useful if you are fetching data directly from a website like PDB or KLIFS.
[8]:
# Let's load a file's content as string (text) to simulate example input data
with open(DATA_PATH / "2itz_chainA_protein.mol2", "r") as f:
text = f.read()
[9]:
DataFrame.from_text(text, "mol2")
[9]:
| atom.id | atom.name | atom.x | atom.y | atom.z | residue.id | residue.name | |
|---|---|---|---|---|---|---|---|
| 0 | 1 | N | 3.5231 | 35.933102 | 15.596400 | 697 | GLU |
| 1 | 2 | H1 | 3.0492 | 36.782101 | 15.323000 | 697 | GLU |
| 2 | 3 | H2 | 3.1546 | 35.156300 | 15.066500 | 697 | GLU |
| 3 | 4 | H3 | 4.5121 | 36.026001 | 15.414000 | 697 | GLU |
| 4 | 5 | CA | 3.3077 | 35.690701 | 17.044600 | 697 | GLU |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 4876 | 4877 | HD2 | 3.5410 | 42.831100 | 42.216801 | 1019 | PRO |
| 4877 | 4878 | HD3 | 1.9924 | 43.615398 | 42.610600 | 1019 | PRO |
| 4878 | 4879 | N | 3.6128 | 43.025902 | 38.110600 | 1020 | GLN |
| 4879 | 4880 | H1 | 4.0174 | 42.840500 | 37.203999 | 1020 | GLN |
| 4880 | 4881 | H2 | 4.1575 | 42.873901 | 38.947399 | 1020 | GLN |
4881 rows × 7 columns
[10]:
DataFrame.from_text(text, "mol2", verbose=True)
[10]:
| atom.id | atom.name | atom.x | atom.y | atom.z | residue.id | residue.name | atom.type | residue.subst_id | residue.subst_name | atom.charge | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | N | 3.5231 | 35.933102 | 15.596400 | 697 | GLU | N.4 | 1 | GLU697 | 1.0 |
| 1 | 2 | H1 | 3.0492 | 36.782101 | 15.323000 | 697 | GLU | H | 1 | GLU697 | 0.0 |
| 2 | 3 | H2 | 3.1546 | 35.156300 | 15.066500 | 697 | GLU | H | 1 | GLU697 | 0.0 |
| 3 | 4 | H3 | 4.5121 | 36.026001 | 15.414000 | 697 | GLU | H | 1 | GLU697 | 0.0 |
| 4 | 5 | CA | 3.3077 | 35.690701 | 17.044600 | 697 | GLU | C.3 | 1 | GLU697 | 0.0 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 4876 | 4877 | HD2 | 3.5410 | 42.831100 | 42.216801 | 1019 | PRO | H | 302 | PRO1019 | 0.0 |
| 4877 | 4878 | HD3 | 1.9924 | 43.615398 | 42.610600 | 1019 | PRO | H | 302 | PRO1019 | 0.0 |
| 4878 | 4879 | N | 3.6128 | 43.025902 | 38.110600 | 1020 | GLN | N.am | 303 | GLN1020 | 0.0 |
| 4879 | 4880 | H1 | 4.0174 | 42.840500 | 37.203999 | 1020 | GLN | H | 303 | GLN1020 | 0.0 |
| 4880 | 4881 | H2 | 4.1575 | 42.873901 | 38.947399 | 1020 | GLN | H | 303 | GLN1020 | 0.0 |
4881 rows × 11 columns
From the pdb format
Check out the pdb format here.
From pdb file: Return a pdb file’s content as DataFrame (ATOM and HETATM entries only).
[11]:
DataFrame.from_file(DATA_PATH / "2itz.pdb")
[11]:
| atom.id | atom.name | atom.x | atom.y | atom.z | residue.id | residue.name | |
|---|---|---|---|---|---|---|---|
| 0 | 1 | N | -67.939003 | -7.787000 | -50.118999 | 697 | GLU |
| 1 | 2 | CA | -67.651001 | -6.991000 | -48.900002 | 697 | GLU |
| 2 | 3 | C | -68.010002 | -5.498000 | -49.028000 | 697 | GLU |
| 3 | 4 | O | -67.916000 | -4.916000 | -50.118999 | 697 | GLU |
| 4 | 5 | CB | -66.192001 | -7.196000 | -48.494999 | 697 | GLU |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 2507 | 2509 | O | -54.306000 | 6.613000 | -8.937000 | 3061 | HOH |
| 2508 | 2510 | O | -54.908001 | 17.349001 | -15.693000 | 3062 | HOH |
| 2509 | 2511 | O | -61.183998 | -5.126000 | -41.009998 | 3063 | HOH |
| 2510 | 2512 | O | -60.660999 | 8.159000 | -21.872000 | 3064 | HOH |
| 2511 | 2513 | O | -69.189003 | 8.275000 | -36.944000 | 3065 | HOH |
2512 rows × 7 columns
In order to get all columns from the input, set verbose=True.
[12]:
DataFrame.from_file(DATA_PATH / "2itz.pdb", verbose=True)
[12]:
| atom.id | atom.name | atom.x | atom.y | atom.z | residue.id | residue.name | record.name | atom.symbol | atom.occupancy | atom.bfactor | atom.alternative_model | structure.chain | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | N | -67.939003 | -7.787000 | -50.118999 | 697 | GLU | ATOM | N | 1.0 | 62.040001 | A | |
| 1 | 2 | CA | -67.651001 | -6.991000 | -48.900002 | 697 | GLU | ATOM | C | 1.0 | 61.730000 | A | |
| 2 | 3 | C | -68.010002 | -5.498000 | -49.028000 | 697 | GLU | ATOM | C | 1.0 | 60.290001 | A | |
| 3 | 4 | O | -67.916000 | -4.916000 | -50.118999 | 697 | GLU | ATOM | O | 1.0 | 61.189999 | A | |
| 4 | 5 | CB | -66.192001 | -7.196000 | -48.494999 | 697 | GLU | ATOM | C | 1.0 | 62.389999 | A | |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 2507 | 2509 | O | -54.306000 | 6.613000 | -8.937000 | 3061 | HOH | HETATM | O | 1.0 | 66.239998 | A | |
| 2508 | 2510 | O | -54.908001 | 17.349001 | -15.693000 | 3062 | HOH | HETATM | O | 1.0 | 58.820000 | A | |
| 2509 | 2511 | O | -61.183998 | -5.126000 | -41.009998 | 3063 | HOH | HETATM | O | 1.0 | 37.119999 | A | |
| 2510 | 2512 | O | -60.660999 | 8.159000 | -21.872000 | 3064 | HOH | HETATM | O | 1.0 | 48.790001 | A | |
| 2511 | 2513 | O | -69.189003 | 8.275000 | -36.944000 | 3065 | HOH | HETATM | O | 1.0 | 44.000000 | A |
2512 rows × 13 columns
From pdb text: Return a pdb string (text) as DataFrame (ATOM and HETATM entries only).
[13]:
# Let's load a file's content as string (text) to simulate example input data
with open(DATA_PATH / "2itz.pdb", "r") as f:
text = f.read()
[14]:
DataFrame.from_text(text, "pdb")
[14]:
| atom.id | atom.name | atom.x | atom.y | atom.z | residue.id | residue.name | |
|---|---|---|---|---|---|---|---|
| 0 | 1 | N | -67.939003 | -7.787000 | -50.118999 | 697 | GLU |
| 1 | 2 | CA | -67.651001 | -6.991000 | -48.900002 | 697 | GLU |
| 2 | 3 | C | -68.010002 | -5.498000 | -49.028000 | 697 | GLU |
| 3 | 4 | O | -67.916000 | -4.916000 | -50.118999 | 697 | GLU |
| 4 | 5 | CB | -66.192001 | -7.196000 | -48.494999 | 697 | GLU |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 2507 | 2509 | O | -54.306000 | 6.613000 | -8.937000 | 3061 | HOH |
| 2508 | 2510 | O | -54.908001 | 17.349001 | -15.693000 | 3062 | HOH |
| 2509 | 2511 | O | -61.183998 | -5.126000 | -41.009998 | 3063 | HOH |
| 2510 | 2512 | O | -60.660999 | 8.159000 | -21.872000 | 3064 | HOH |
| 2511 | 2513 | O | -69.189003 | 8.275000 | -36.944000 | 3065 | HOH |
2512 rows × 7 columns
[15]:
DataFrame.from_text(text, "pdb", verbose=True)
[15]:
| atom.id | atom.name | atom.x | atom.y | atom.z | residue.id | residue.name | record.name | atom.symbol | atom.occupancy | atom.bfactor | atom.alternative_model | structure.chain | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | N | -67.939003 | -7.787000 | -50.118999 | 697 | GLU | ATOM | N | 1.0 | 62.040001 | A | |
| 1 | 2 | CA | -67.651001 | -6.991000 | -48.900002 | 697 | GLU | ATOM | C | 1.0 | 61.730000 | A | |
| 2 | 3 | C | -68.010002 | -5.498000 | -49.028000 | 697 | GLU | ATOM | C | 1.0 | 60.290001 | A | |
| 3 | 4 | O | -67.916000 | -4.916000 | -50.118999 | 697 | GLU | ATOM | O | 1.0 | 61.189999 | A | |
| 4 | 5 | CB | -66.192001 | -7.196000 | -48.494999 | 697 | GLU | ATOM | C | 1.0 | 62.389999 | A | |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 2507 | 2509 | O | -54.306000 | 6.613000 | -8.937000 | 3061 | HOH | HETATM | O | 1.0 | 66.239998 | A | |
| 2508 | 2510 | O | -54.908001 | 17.349001 | -15.693000 | 3062 | HOH | HETATM | O | 1.0 | 58.820000 | A | |
| 2509 | 2511 | O | -61.183998 | -5.126000 | -41.009998 | 3063 | HOH | HETATM | O | 1.0 | 37.119999 | A | |
| 2510 | 2512 | O | -60.660999 | 8.159000 | -21.872000 | 3064 | HOH | HETATM | O | 1.0 | 48.790001 | A | |
| 2511 | 2513 | O | -69.189003 | 8.275000 | -36.944000 | 3065 | HOH | HETATM | O | 1.0 | 44.000000 | A |
2512 rows × 13 columns
Column types:
[16]:
DataFrame.from_file(DATA_PATH / "2itz_chainA_protein.mol2", verbose=True).dtypes
[16]:
atom.id int32
atom.name string
atom.x float32
atom.y float32
atom.z float32
residue.id string
residue.name string
atom.type string
residue.subst_id Int64
residue.subst_name string
atom.charge float32
dtype: object
[17]:
DataFrame.from_file(DATA_PATH / "2itz.pdb", verbose=True).dtypes
[17]:
atom.id int32
atom.name string
atom.x float32
atom.y float32
atom.z float32
residue.id string
residue.name string
record.name string
atom.symbol string
atom.occupancy float32
atom.bfactor float32
atom.alternative_model string
structure.chain string
dtype: object
Load structural data as rdkit molecule
Note: rdkit is a cheminformatics toolkit that focuses on working with small molecules. Technically you can load protein structures as rdkit molecule, however we recommend to use this data structure for small molecules only.
From the mol2 format
From mol2 files: Return a mol2 file’s content as DataFrame (mol2 files can have 9 (default) or 10 columns).
[18]:
Rdkit.from_file(DATA_PATH / "2itz_chainA_ligand.mol2")
[18]:
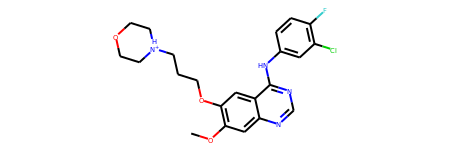
[19]:
Rdkit.from_file(DATA_PATH / "2itz_chainA_ligand.mol2", compute2d=False)
[19]:
From mol2 text: Return a mol2 string (text) as DataFrame. This functionality is useful if you are fetching data directly from a website like KLIFS.
[20]:
# Let's load a file's content as string (text) to simulate example input data
with open(DATA_PATH / "2itz_chainA_ligand.mol2", "r") as f:
text = f.read()
[21]:
Rdkit.from_text(text, "mol2")
[21]:
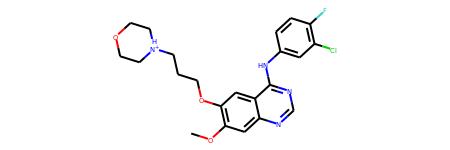
Load structural data as biopython’s Structure objects
[22]:
structure = Biopython.from_file(DATA_PATH / "2itz.pdb")
structure
[22]:
<Structure id=>
[23]:
type(structure)
[23]:
Bio.PDB.Structure.Structure
[24]:
print(f"Number of residues: {len(list(structure.get_residues()))}")
Number of residues: 370