The opencadd.structure.pocket module
Let’s walk through the functionalities offered in the opencadd.structure.pocket module.
[1]:
%load_ext autoreload
%autoreload 2
[2]:
from pathlib import Path
import pandas as pd
from opencadd.databases.klifs import setup_remote, setup_local
from opencadd.structure.pocket import Pocket, PocketKlifs, PocketViewer
[3]:
HERE = Path(_dh[-1])
KLIFS_DOWNLOAD_PATH = HERE / "../../opencadd/tests/data/klifs"
Get example protein structure and pocket residues
[4]:
from opencadd.databases.klifs import setup_remote
KLIFS_REMOTE = setup_remote()
Fetch protein structure file content
First of all, we download structural data for an example protein kinase from the KLIFS database (some pocket residues are missing):
https://klifs.net/details.php?structure_id=12347
[5]:
structure_klifs_id = 12347
[6]:
text = KLIFS_REMOTE.coordinates.to_text(structure_klifs_id, extension="pdb")
Fetch pocket residues (or use your own pocket residues)
[7]:
pocket_residues = KLIFS_REMOTE.pockets.by_structure_klifs_id(structure_klifs_id)
pocket_residues
[7]:
| residue.klifs_id | residue.id | residue.klifs_region_id | residue.klifs_region | residue.klifs_color | |
|---|---|---|---|---|---|
| 0 | 1 | 461 | I.1 | I | khaki |
| 1 | 2 | 462 | I.2 | I | khaki |
| 2 | 3 | 463 | I.3 | I | khaki |
| 3 | 4 | _ | g.l.4 | g.l | green |
| 4 | 5 | _ | g.l.5 | g.l | green |
| ... | ... | ... | ... | ... | ... |
| 80 | 81 | 594 | xDFG.81 | xDFG | cornflowerblue |
| 81 | 82 | 595 | xDFG.82 | xDFG | cornflowerblue |
| 82 | 83 | _ | xDFG.83 | xDFG | cornflowerblue |
| 83 | 84 | _ | a.l.84 | a.l | cornflowerblue |
| 84 | 85 | _ | a.l.85 | a.l | cornflowerblue |
85 rows × 5 columns
The variables pocket_residue_ids and pocket_residue_ixs contain the list of residue PDB IDs and residue indices (derived from KLIFS sequence- and structure-based alignment). We will need this pocket information in the next step where we want to set up a pocket from opencadd’s Pocket class.
[8]:
pocket_residue_ids = pocket_residues["residue.id"].to_list()
print("Pocket residue PDB IDs:")
print(*pocket_residue_ids)
pocket_residue_ixs = pocket_residues["residue.klifs_id"].to_list()
print("Pocket residue (KLIFS) indices:")
print(*pocket_residue_ixs)
Pocket residue PDB IDs:
461 462 463 _ _ _ _ 468 469 470 471 472 473 480 481 482 483 484 485 497 498 499 500 501 502 503 504 505 506 507 508 509 511 512 513 514 515 516 517 518 519 526 527 528 529 530 531 532 533 534 535 536 537 538 539 540 541 542 543 566 567 568 569 570 571 572 573 574 575 576 577 578 579 580 581 582 583 584 592 593 594 595 _ _ _
Pocket residue (KLIFS) indices:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85
Pocket (Pocket class)
The Pocket class currently holds the following attributes/properties:
name: Protein/pocket namefilepath: Path to file with structural protein datacentroid: Centroids of all pocket residues’ CA atomssubpockets: Subpockets defined based on a set of anchor residues eachregions: User-defined regions that are of importance for the protein/pocketanchor_residues: Anchor residues to define one or more subpockets
Initialize pocket
We initialize the pocket with the following parameters:
Protein structure data
Protein/pocket name
Pocket residues PDB IDs
Pocket residue indices (optionally), e.g. for the pocket alignment IDs
[9]:
pocket = Pocket.from_text(
text,
"pdb",
pocket_residue_ids,
pocket_residue_ixs,
name=structure_klifs_id
)
Let’s take a look at key Pocket class attributes/properties after initialization.
Pocket residues
All residue PDB IDs that cannot be cast to an integer are set to None.
[10]:
pocket.residues
[10]:
| residue.id | residue.ix | |
|---|---|---|
| 0 | 461 | 1 |
| 1 | 462 | 2 |
| 2 | 463 | 3 |
| 3 | <NA> | 4 |
| 4 | <NA> | 5 |
| ... | ... | ... |
| 80 | 594 | 81 |
| 81 | 595 | 82 |
| 82 | <NA> | 83 |
| 83 | <NA> | 84 |
| 84 | <NA> | 85 |
85 rows × 2 columns
[11]:
print(*pocket._residue_ids)
461 462 463 None None None None 468 469 470 471 472 473 480 481 482 483 484 485 497 498 499 500 501 502 503 504 505 506 507 508 509 511 512 513 514 515 516 517 518 519 526 527 528 529 530 531 532 533 534 535 536 537 538 539 540 541 542 543 566 567 568 569 570 571 572 573 574 575 576 577 578 579 580 581 582 583 584 592 593 594 595 None None None
[12]:
print(*pocket._residue_ixs)
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85
Pocket centroids
[13]:
pocket.center
[13]:
array([ 0.8315384, 21.615948 , 36.450153 ], dtype=float32)
Pocket data (atoms as DataFrame)
[14]:
pocket.data
[14]:
| atom.id | atom.name | atom.x | atom.y | atom.z | residue.id | residue.name | |
|---|---|---|---|---|---|---|---|
| 73 | 74 | N | 9.014 | 18.239000 | 51.860001 | 461 | GLN |
| 74 | 75 | CA | 8.811 | 16.811001 | 51.655998 | 461 | GLN |
| 75 | 76 | C | 8.559 | 16.492001 | 50.186001 | 461 | GLN |
| 76 | 77 | O | 7.851 | 17.224001 | 49.491001 | 461 | GLN |
| 77 | 78 | CB | 7.630 | 16.325001 | 52.500999 | 461 | GLN |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 1015 | 1016 | CD1 | -0.042 | 21.966000 | 31.419001 | 595 | PHE |
| 1016 | 1017 | CD2 | 1.547 | 22.907000 | 29.912001 | 595 | PHE |
| 1017 | 1018 | CE1 | -0.337 | 23.228001 | 31.899000 | 595 | PHE |
| 1018 | 1019 | CE2 | 1.255 | 24.172001 | 30.389000 | 595 | PHE |
| 1019 | 1020 | CZ | 0.311 | 24.332001 | 31.384001 | 595 | PHE |
577 rows × 7 columns
Add subpockets
Next, we can add subpockets one-by-one to the pocket. For each subpocket we define the following:
Subpocket name
Residue PDB IDs OR residue indices (e.g. alignment indices) of all anchor residues, i.e. the residues determining the subpocket center (centroid of all anchor residues’ CA atoms)
Subpocket color
The class method add_subpocket uses the Subpocket class to set up subpockets.
[15]:
pocket.add_subpocket("hinge_region", anchor_residue_ixs=[16, 47, 80], color="magenta")
pocket.add_subpocket("dfg_region", anchor_residue_ixs=[19, 24, 81], color="cornflowerblue")
pocket.add_subpocket("front_pocket", anchor_residue_ixs=[10, 48, 72], color="cyan")
Using the Pocket’s property subpockets, we get an overview of all specified subpockets.
[16]:
pocket.subpockets
[16]:
| subpocket.name | subpocket.color | subpocket.center | |
|---|---|---|---|
| 0 | hinge_region | magenta | [2.4566665, 22.592667, 41.674] |
| 1 | dfg_region | cornflowerblue | [8.460667, 20.395666, 33.809666] |
| 2 | front_pocket | cyan | [0.6393334, 16.937, 39.594666] |
Using the Pocket‘s property anchor_residues, we get an overview of all subpockets’ anchor residues.
[17]:
pocket.anchor_residues
[17]:
| subpocket.name | anchor_residue.color | anchor_residue.id | anchor_residue.id_alternative | anchor_residue.ix | anchor_residue.center | |
|---|---|---|---|---|---|---|
| 0 | hinge_region | magenta | 482 | None | 16 | [8.327, 22.785, 43.461] |
| 1 | hinge_region | magenta | 531 | None | 47 | [-0.001, 24.108, 46.55] |
| 2 | hinge_region | magenta | 593 | None | 80 | [-0.956, 20.885, 35.011] |
| 3 | dfg_region | cornflowerblue | 485 | None | 19 | [15.03, 17.46, 38.074] |
| 4 | dfg_region | cornflowerblue | 501 | None | 24 | [8.524, 25.293, 29.217] |
| 5 | dfg_region | cornflowerblue | 594 | None | 81 | [1.828, 18.434, 34.138] |
| 6 | front_pocket | cyan | 470 | None | 10 | [11.026, 15.031, 41.426] |
| 7 | front_pocket | cyan | 532 | None | 48 | [-3.527, 22.678, 46.33] |
| 8 | front_pocket | cyan | 578 | None | 72 | [-5.581, 13.102, 31.028] |
Subpockets are calculated based on so class anchor residues, defined each in an AnchorResidue class. Subpocket centers are the centroids of all anchor residues’ centers (i.e. normally the CA atoms).
If the anchor residue’s CA atom is available in the input structure is available, its coordinates are defined as the anchor residues center.
If the anchor residue’s CA atom is missing in a structure, alternative anchors are chosen if possible: If the residue CA atoms before and after the input anchor residue are available, their CA atoms’ centroid is chosen.
If only one of the neighboring residues’ CA atoms is available, that single CA atoms is chosen.
If none of the anchor residue’s and neighboring residues’ CA atoms is available, no anchor residue center is defined.
The determination of anchor residues depends on the CA atom availablity of the user-defined anchor residue as well as the residue before and after.
Add regions
The Pocket class also allows to specify pocket regions, normally used to store key regions, such as the hinge region or the catalytic loop in kinases. This information can be used for pocket visualization.
The class method add_regions uses the Regions class to set up regions.
[18]:
pocket.add_region("hinge", residue_ixs=[46, 47, 48], color="magenta")
pocket.add_region("linker", residue_ixs=[49, 50, 51, 52], color="cyan")
pocket.add_region("xDFG", residue_ixs=[80, 81, 82, 83], color="cornflowerblue")
[19]:
pocket.regions
[19]:
| region.name | region.color | residue.id | residue.ix | |
|---|---|---|---|---|
| 0 | hinge | magenta | 530 | 46 |
| 1 | hinge | magenta | 531 | 47 |
| 2 | hinge | magenta | 532 | 48 |
| 3 | linker | cyan | 533 | 49 |
| 4 | linker | cyan | 534 | 50 |
| 5 | linker | cyan | 535 | 51 |
| 6 | linker | cyan | 536 | 52 |
| 7 | xDFG | cornflowerblue | 593 | 80 |
| 8 | xDFG | cornflowerblue | 594 | 81 |
| 9 | xDFG | cornflowerblue | 595 | 82 |
| 10 | xDFG | cornflowerblue | <NA> | 83 |
Visualize pocket
Besides the pocket, we also want to visualize the co-crystallized ligand (if any), so let’s fetch the ligand Expo ID.
[20]:
ligand_expo_id = KLIFS_REMOTE.structures.by_structure_klifs_id(structure_klifs_id)["ligand.expo_id"][0]
[21]:
viewer = PocketViewer()
viewer.add_pocket(pocket, ligand_expo_id=ligand_expo_id)
[22]:
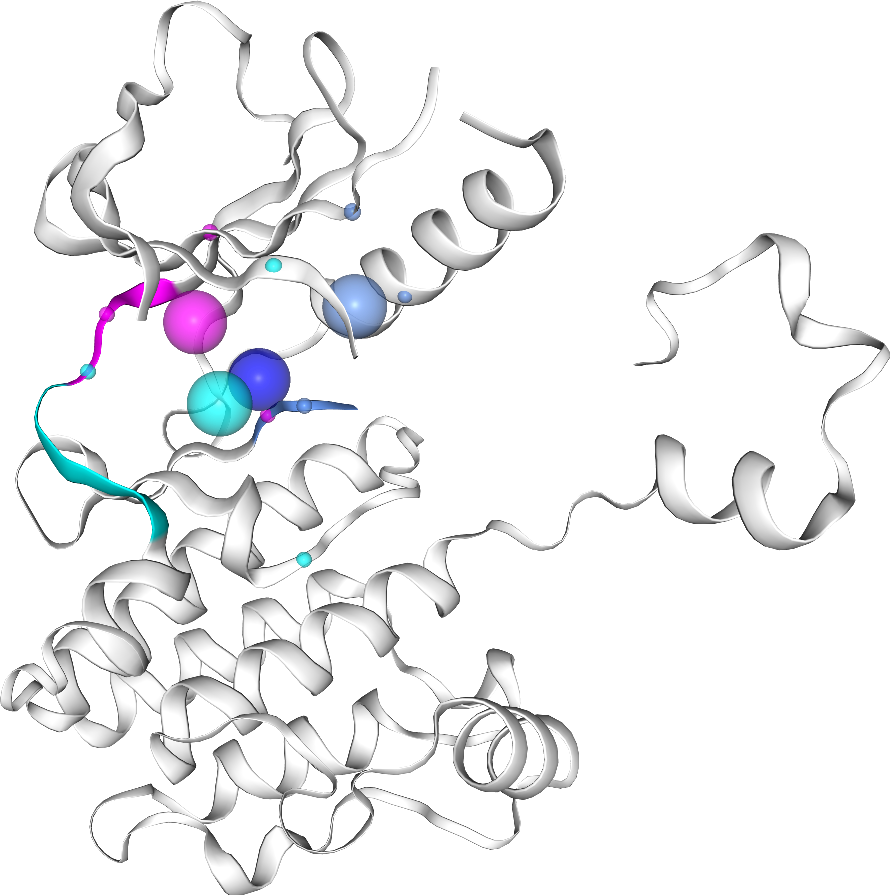
viewer.viewer.render_image(trim=True, factor=2, transparent=True),
[22]:
(Image(value=b'', width='99%'),)
[23]:
# Static output
viewer.viewer._display_image()
[23]:
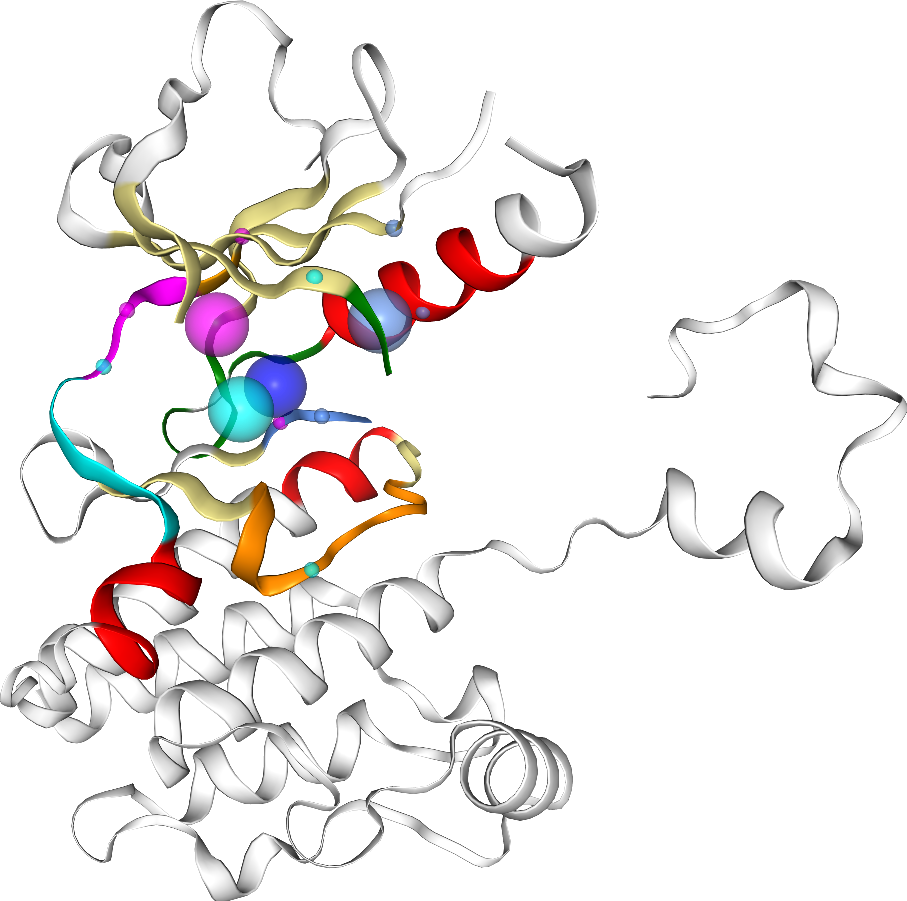
KLIFS pocket (PocketKlifs class)
The PocketKlifs class is a child of the Pocket class, setting the kinase pocket regions as defined by KLIFS.
Figure 1: Kinase pocket regions as defined by KLIFS (taken from the KLIFS publication)
Define subpockets (name and color) based on user-defined KLIFS residue IDs.
[24]:
subpockets = {
"anchor_residue.klifs_ids": [[16, 47, 80], [19, 24, 81], [10, 48, 72]],
"subpocket.name": ["hinge_region", "dfg_region", "front_pocket"],
"subpocket.color": ["magenta", "cornflowerblue", "cyan"]
}
subpockets = pd.DataFrame(subpockets)
subpockets
[24]:
| anchor_residue.klifs_ids | subpocket.name | subpocket.color | |
|---|---|---|---|
| 0 | [16, 47, 80] | hinge_region | magenta |
| 1 | [19, 24, 81] | dfg_region | cornflowerblue |
| 2 | [10, 48, 72] | front_pocket | cyan |
Initialize KLIFS pocket
… from remote KLIFS session
[25]:
kinase_pocket = PocketKlifs.from_structure_klifs_id(structure_klifs_id, subpockets)
This will internally, initiate a remote KLIFS session to fetch the relevant data. If you have a remote KLIFS session already initialized, you can also use it directly.
[26]:
kinase_pocket = PocketKlifs.from_structure_klifs_id(
structure_klifs_id,
subpockets,
klifs_session=KLIFS_REMOTE
)
… from local KLIFS session
Use an example local KLIFS dataset.
[27]:
KLIFS_LOCAL = setup_local(KLIFS_DOWNLOAD_PATH)
… based on mol2 files
[28]:
kinase_pocket = PocketKlifs.from_structure_klifs_id(
structure_klifs_id,
subpockets,
extension="mol2",
klifs_session=KLIFS_LOCAL
)
Suspicious residue ID: _0 (from QH1_0)
… based on pdb files
[29]:
kinase_pocket = PocketKlifs.from_structure_klifs_id(
structure_klifs_id,
subpockets,
extension="pdb",
klifs_session=KLIFS_LOCAL
)
Visualize pocket with all KLIFS-defined regions
[30]:
viewer = PocketViewer()
viewer.add_pocket(kinase_pocket)
[31]:
viewer.viewer.render_image(trim=True, factor=2, transparent=True),
[31]:
(Image(value=b'', width='99%'),)
[32]:
# Static output
viewer.viewer._display_image()
[32]: